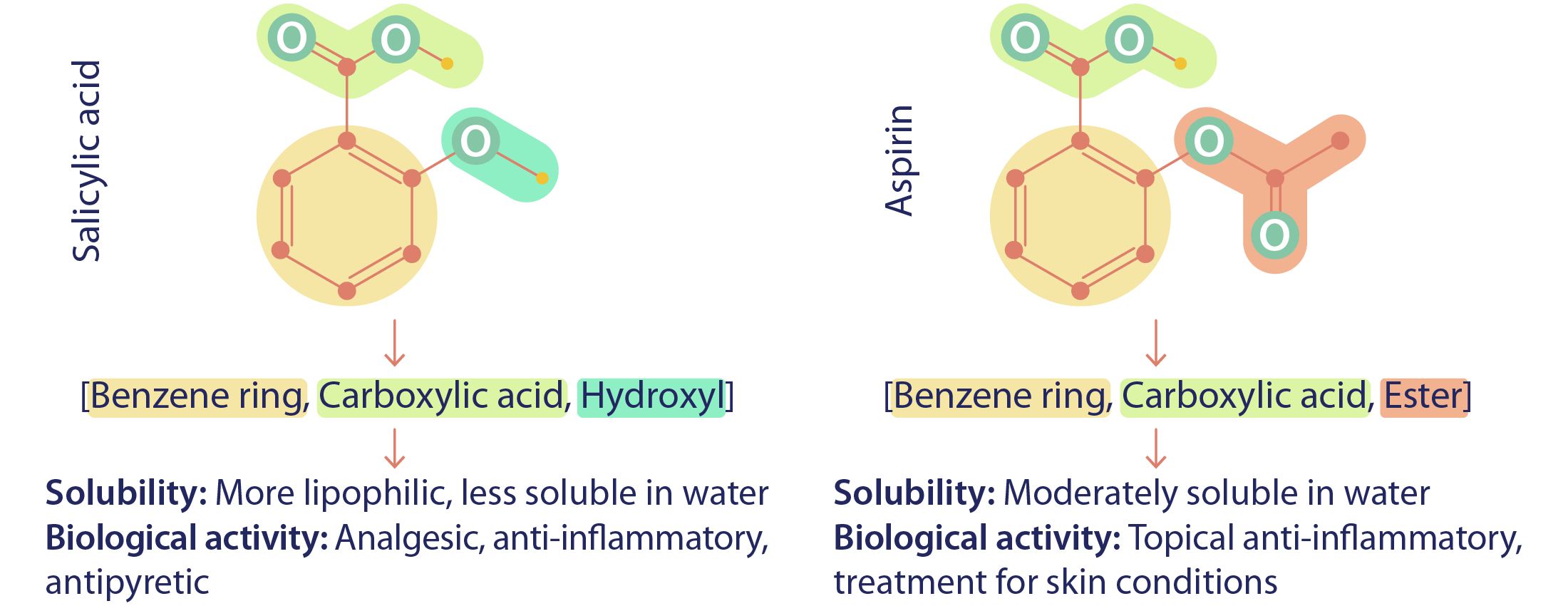
The exploration of chemical compounds often hinges on understanding the unique roles that functional groups play in determining molecular properties and behaviors, as even a slight change in a functional group can lead to significant differences in properties. For example, while aspirin and salicylic acid are structurally similar, the only difference lies in the substitution of the hydroxyl group (OH) in salicylic acid with an ester (COO) in aspirin, transforming a skincare compound into an effective drug. However, traditional molecular representations, such as SMILES, treat atoms as isolated entities, lacking the nuanced insight provided by functional group interactions.
Based on this insight, we designed FARM (Functional Group-Aware Representations for Small Molecules). The key innovation of FARM lies in its functional group-aware tokenization, which incorporates functional group information directly into the representations. This strategic reduction in tokenization granularity, intentionally interfaced with key drivers of functional properties (i.e., functional groups), enhances the model's understanding of chemical language, expands the chemical lexicon, bridges the gap between SMILES and natural language, and ultimately advances the model's capacity to predict molecular properties.
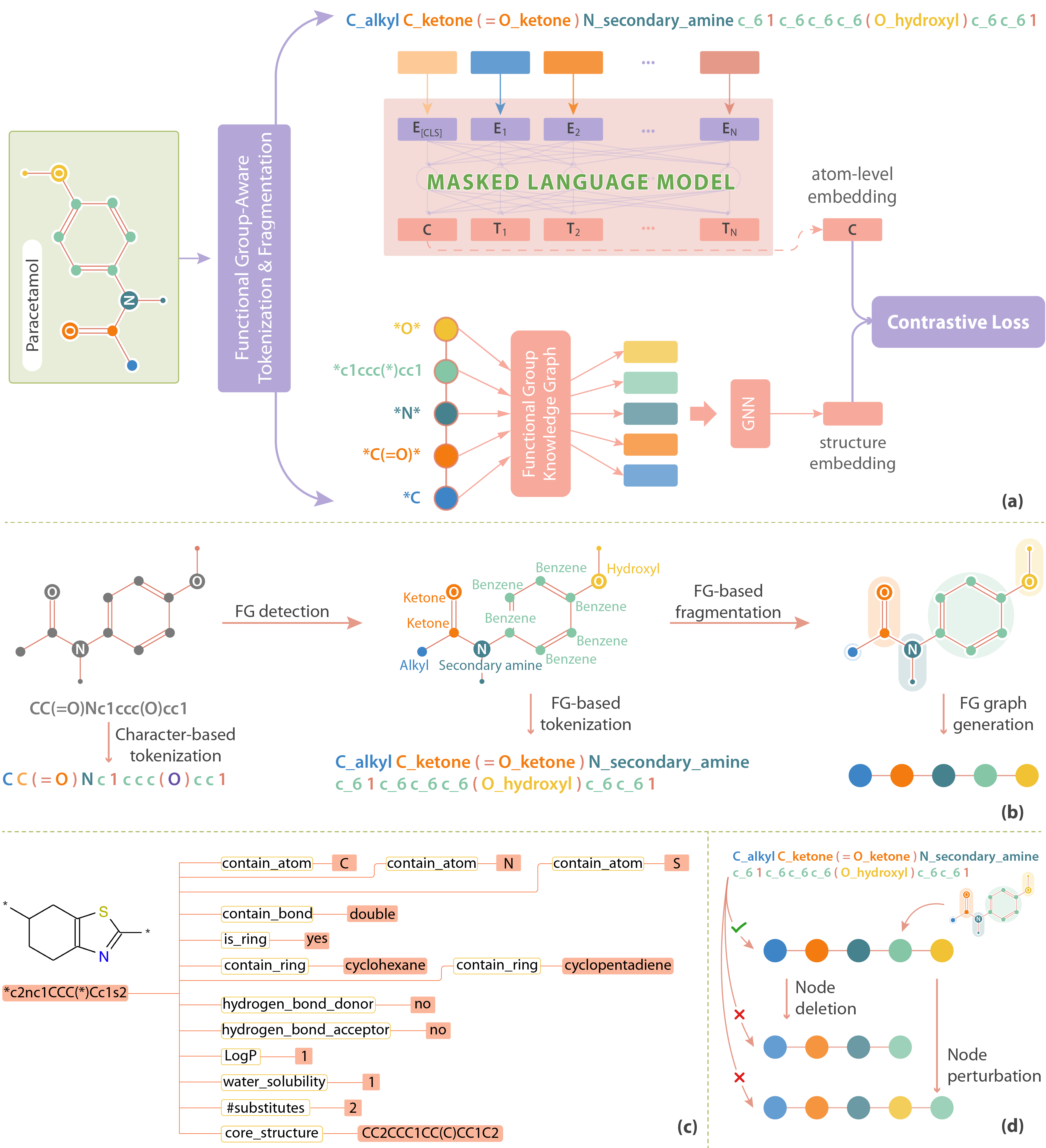
Negative transfer occurs when a model trained on one domain or task struggles to generalize effectively to another due to differences in the underlying patterns. In molecular modeling, using only individual atom tokens without functional group information can lead to poor learning because the model fails to capture the full context of molecular behavior. The small, simplistic vocabulary of atom types, typically around 100 tokens, limits the model’s ability to understand complex chemical properties, often resulting in incorrect generalizations or degraded performance across different molecules and tasks.
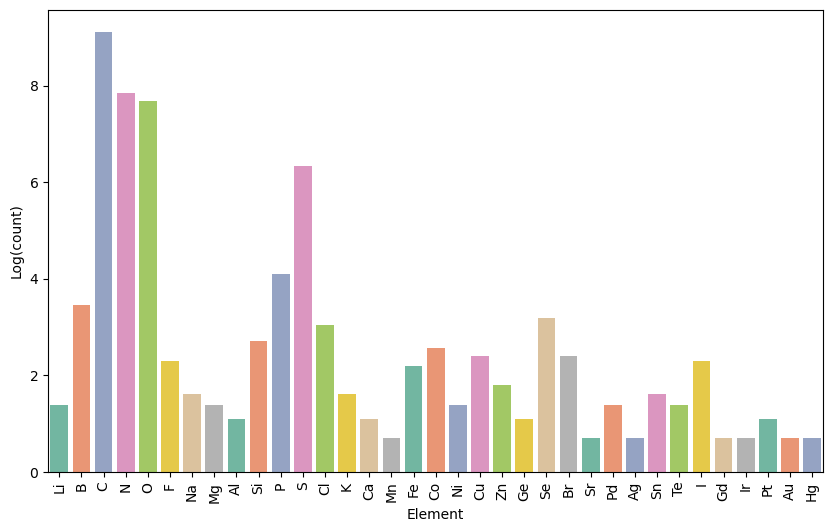
To address this, our FG-aware tokenization expands the vocabulary from 93 tokens to approximately 14,741 tokens by incorporating functional group information. While this significantly increases the complexity of the model, making training slower and harder to converge, it also prevents negative transfer by enabling the model to learn richer and more meaningful chemical semantics. This larger, more nuanced vocabulary allows the model to better capture the functional roles of atoms within molecules, improving its ability to generalize across tasks and ultimately leading to more accurate molecular representations.
We rigorously evaluate FARM on the MoleculeNet dataset, where it achieves state-of-the-art performance on 10 out of 12 tasks. These results highlight FARM’s potential to improve molecular representation learning, with promising applications in drug discovery and pharmaceutical research. Below is an analysis of molecule embeddings generated by FARM:
Masked language prediction model (BERT) for atom-level representation learning
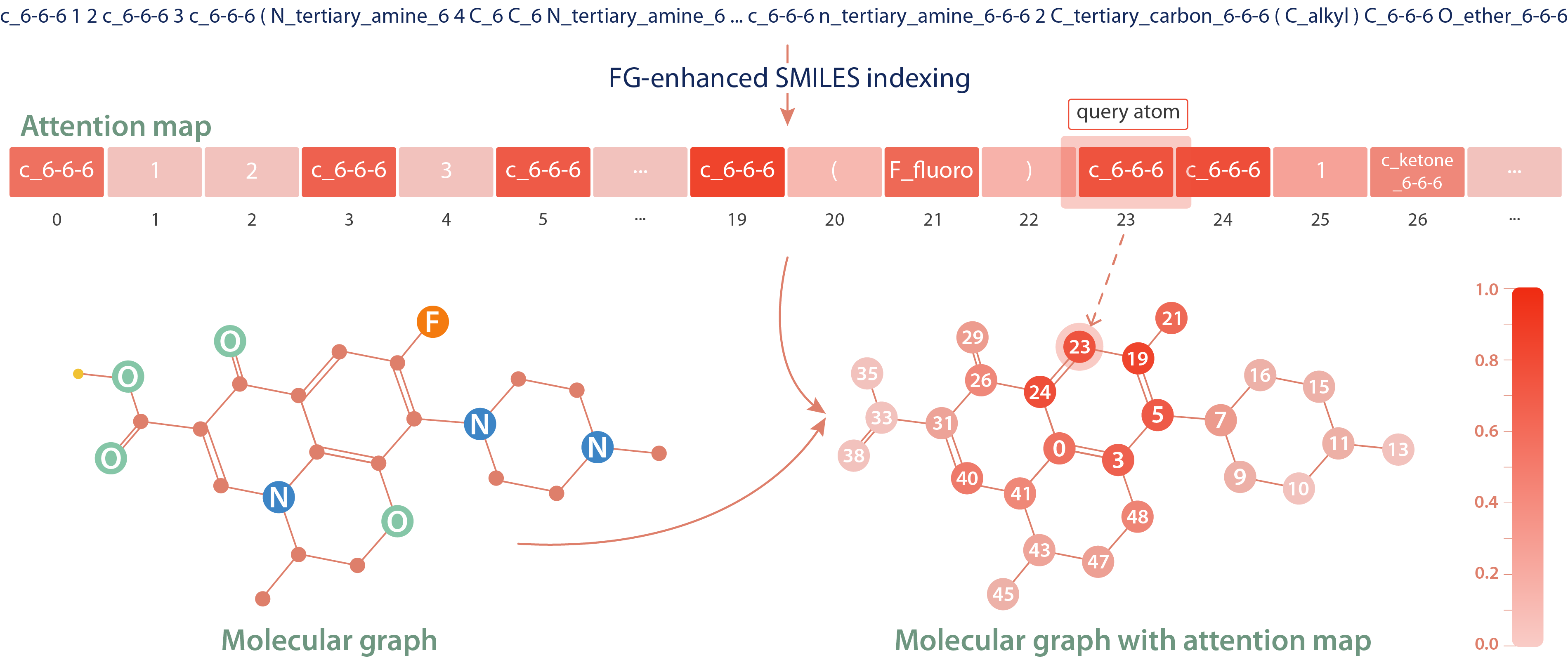
We examine the attention mechanism of the BERT model trained with FG-enhanced SMILES by visualizing the attention scores for a query atom. The attention map reveals that the model pays more attention to atoms that are strongly connected to the query atom than to those that are merely adjacent in the SMILES string. In detail, the query atom at position 23 shows higher attention to the atom at position 0, which is part of the same ring, rather than to the atom at position 26, which is closer in the SMILES string but not directly connected. This demonstrates that the model effectively learns the syntax and semantics of SMILES, capturing the underlying molecular structure rather than merely relying on the linear sequence of SMILES.
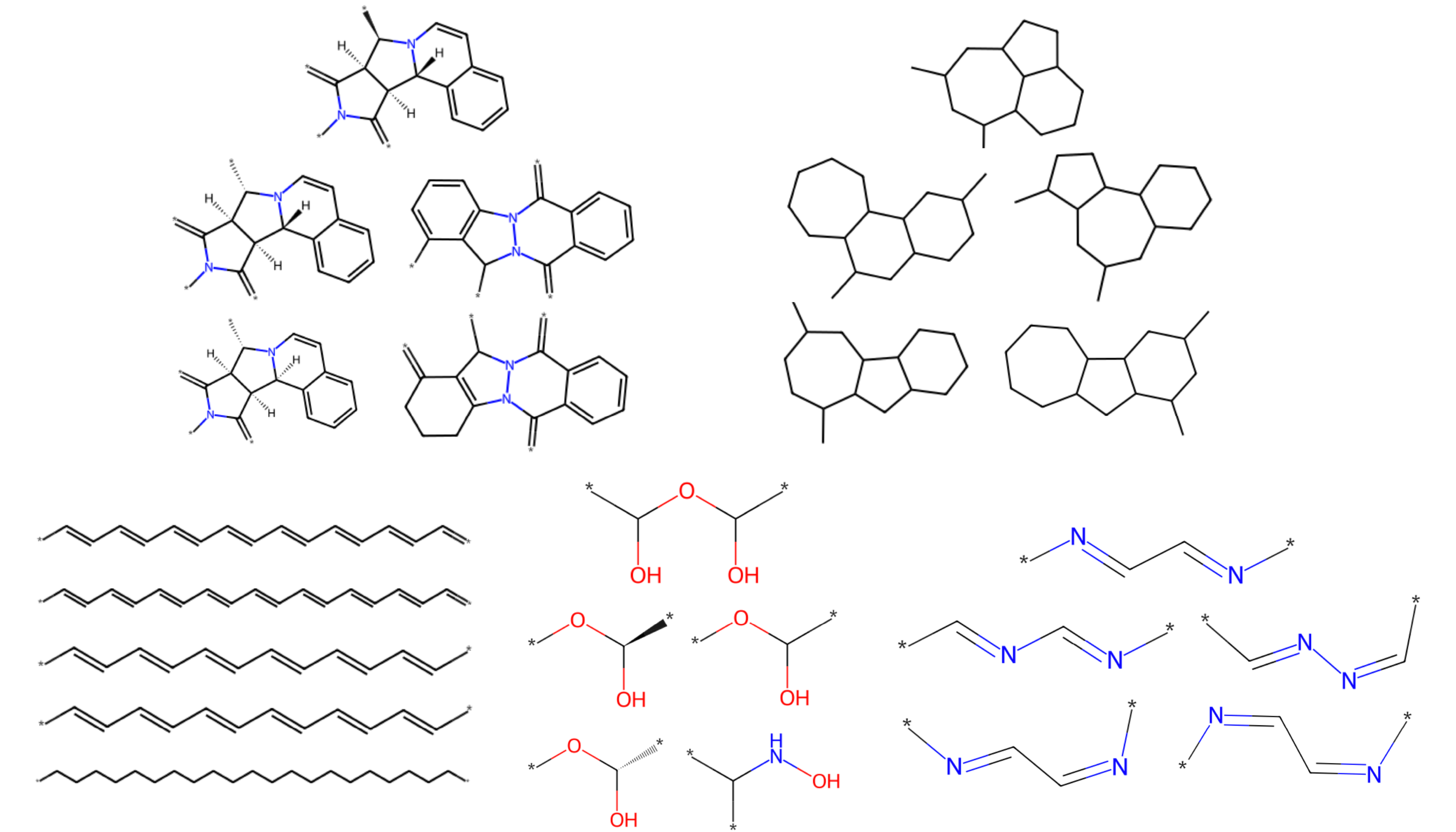
FG knowledge graph for functional group embeddings
To assess the quality of the learned embeddings, we randomly sampled clusters of five closely related embedding vectors and analyzed their arrangement in the embedding space. The results demonstrate that similar FGs tend to cluster together, indicating that the FG knowledge graph embeddings effectively capture relationships between FGs.
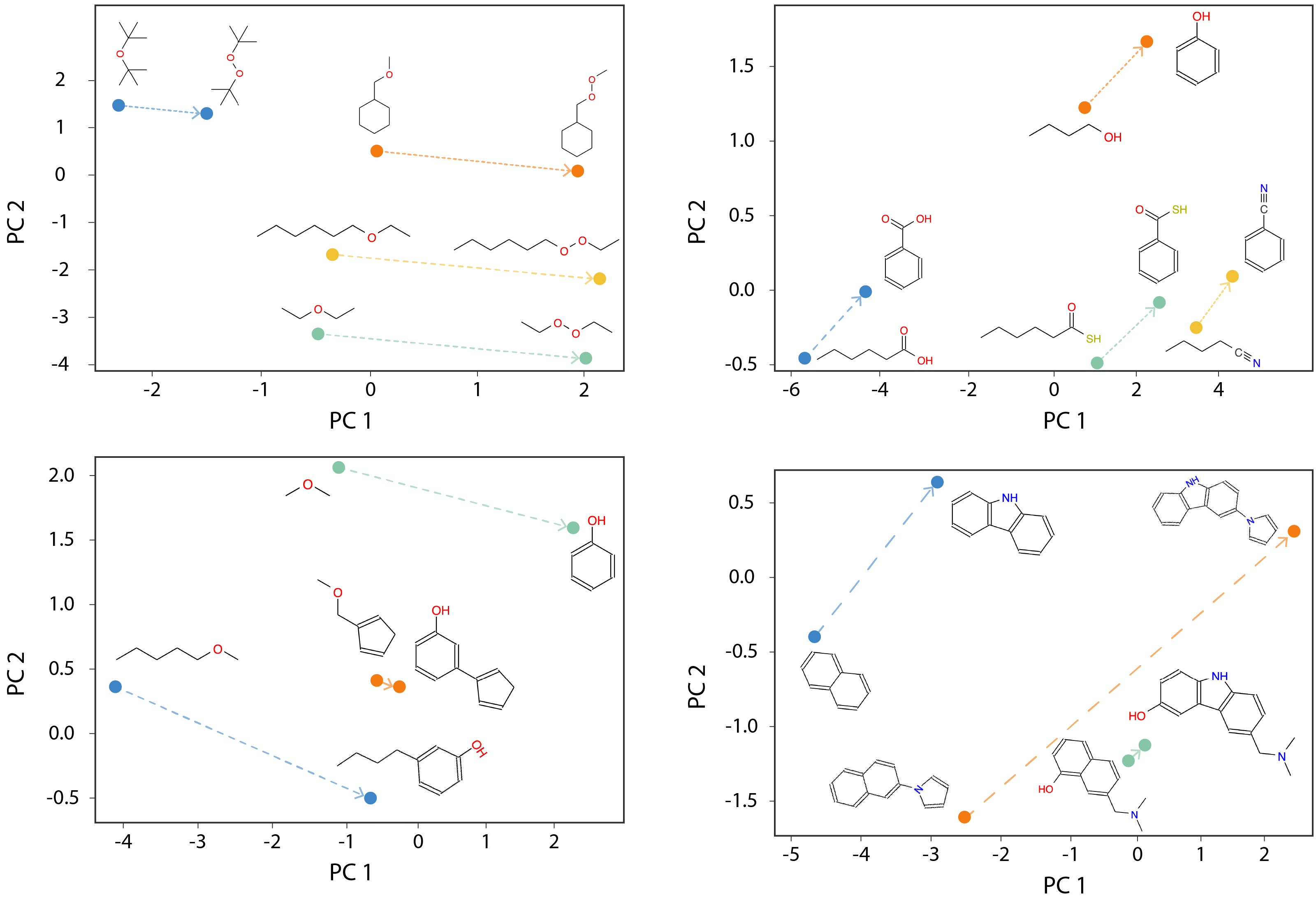
GCN link prediction model for molecular structure learning
Inspired by word pair analogy tasks in NLP, we replace one functional group in a molecule with another and observe consistent parallel results across different molecules in the molecular structure embedding space. This demonstrates the model’s ability to effectively capture and preserve chemical analogies, highlighting its robustness in learning and representing molecular structures.
While FARM shows strong performance, there are two main limitations that should be addressed in future work. First, the current model does not incorporate a full 3D molecular representation, which is critical for capturing stereochemistry and spatial configurations that affect molecular properties. Incorporating 3D information Yan et al. (2024) could further enhance the model’s predictions. Second, the model faces challenges when dealing with rare fused ring systems due to out-of-vocabulary issues. A potential solution to this limitation is to extend the training dataset, covering a broader portion of chemical space to include more diverse and complex molecular structures.
Looking ahead, our ultimate goal is to develop a pre-trained atom embedding that parallels the capabilities of pre-trained word embeddings in NLP. This would enable a richer and more nuanced understanding of molecular properties and behaviors at the atomic level. Similarly, we aim to achieve molecule-level representations that are as expressive and versatile as sentence-level embeddings in NLP, capturing both local and global molecular features. By bridging the gap between atom-wise embeddings and holistic molecule representations, FARM paves the way for more accurate, generalizable molecular predictions across a variety of tasks.
@article{nguyen2024farm,
title={{FARM}: Functional Group-Aware Representations for Small Molecules},
author={Thao Nguyen and Kuan-Hao Huang and Ge Liu and Martin D. Burke and Ying Diao and Heng Ji},
journal={arXiv preprint arXiv:2410.02082},
year={2024}
}